Chapter 2 Introduction to GWAS, Ancestry, and Local Ancestry Inference
2.1 Basic Genetics
The human genome consists of 23 pairs of chromosomes, each of which contains millions of nucleotide base pairs joined together in a double helix. The nucleotides adenine (A) and thymine (T) always pair with each other, as do cytosine (C) and guanine (G). At any point along a person’s DNA strand, the sequence of A’s, T’s, C’s, and G’s dictates characteristics of that person. When comparing any two human genomes, the entire strands of nucleotides will be about 99% identical. However, there are small portions of the genome where the sequences vary from person to person, and these places are called genetic variants. Different nucleotide sequences in these areas are what cause variation in the human population.
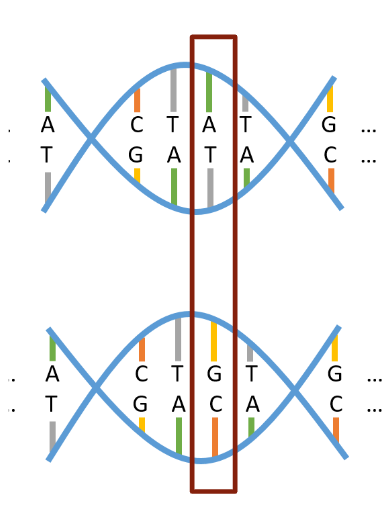
Figure 1: This image depicts segments of two copies of a person’s chromosome, representing one received from each parent. The outlined section represents a SNV, showing a single position along the strands where different alleles (A/T vs. G/C) were received from the two parents. Image sourced from Prof. Grinde’s STAT 494 class notes.
When humans are conceived, they receive one copy of all 23 chromosomes from their mother, and one copy from their father, as depicted in Figure 1. Thus, when considering a single-nucleotide genetic variant (SNV) at a particular position on any chromosome, it is the case that a person will have received one allele (A/T, G/C, etc.) from each parent at that position. These two alleles together carry the meaning of that SNV, potentially in the form of an observable trait. Most SNVs are considered biallelic, meaning only two of the four total base pair combinations are possible to be inherited as alleles at a SNV’s particular genomic position. The pair that occurs more commonly across the human population at a SNV is referred to as the major allele, and the less common pair is referred to as the minor allele. Whether a person received 0, 1, or 2 minor alleles from their parents at a given position determines their minor allele frequency (MAF) at that spot. If the overall MAF of a given SNV is greater than 1% across the human population, it may instead be referred to as a single-nucleotide polymorphism (SNP). It is data on individuals’ MAFs at SNPs in particular that is commonly used as the backbone of genetic studies.
2.2 GWAS and Why Ancestry Matters
In order to learn about how various SNPs are linked to observable human traits, geneticists often conduct what are called genome-wide association studies (GWAS). These studies involve gathering genetic and observational data on many individuals, some of whom may possess a certain trait of interest. After acquiring the exact sequence of subjects’ genomes, experimenters sift through the sequences for SNPs and record subjects’ MAFs at each one. Then, regression analysis can be performed to find correlations between individuals’ MAFs at various SNPs and their observed traits. This helps experimenters draw conclusions about which SNPs are universally important in determining those traits.
Ancestry, however, can often be a confounding factor that muddles the true relationship between SNPs and observed traits during GWAS, as depicted in Figure 2. Consider, for example, a study attempting to determine which SNPs cause brown hair, but without accounting for German ancestry as a possible confounder. A genotype with SNPs resulting from a German pedigree might show a strong correlation with brown hair if people of German descent are more likely to have brown hair in general. However, concluding that those particular SNPs cause brown hair would be erroneous, since it was really the fact that the people with those certain SNP characteristics were of German origin that caused them to have brown hair. Thus, gathering information about subjects’ ancestries is a vital part of GWAS, in that including such data in the regression models helps correct for this type of confounding effect.
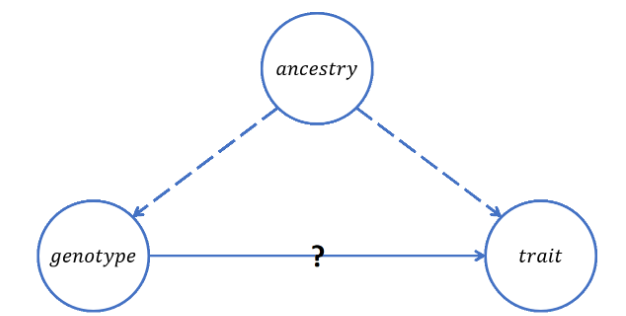
Figure 2: This is a DAG depicting the confounding effect of ancestry in GWAS. Doubt can be cast on a potential relationship between a genotype and a trait if ancestry has a true causal relationship with both.Image sourced from Prof. Grinde’s STAT 494 class notes.
2.3 Project Motivations & Local Ancestry Inference
Our Statistical Genetics course has provided us with a diverse sampling of methods used for genome wide association studies. For our project, we wanted to use this tool set of methodologies to expand on themes of ancestry and classification by honing in on local ancestry inference (LAI). That is, we wanted to see if we could infer the ancestral origins of certain segments of an individual’s genome based on the presence, or absence, of genetic variants of interest.
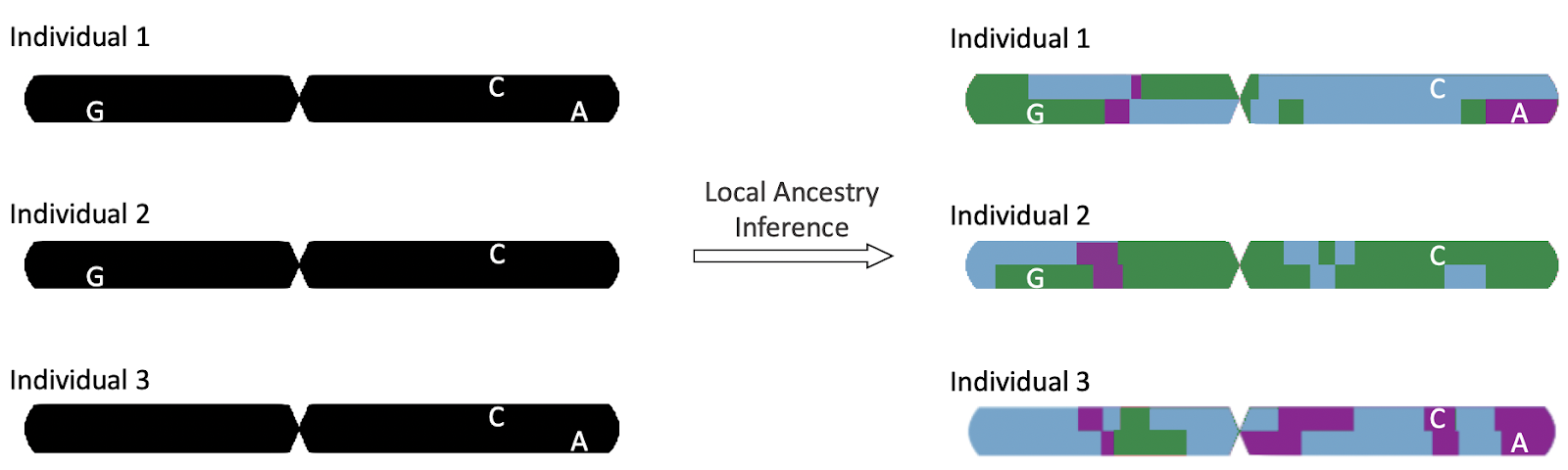
Figure 3: Local ancestry inference can allow researchers to find the allele ancestries at each locus of an individual’s genome, denoted by blocks of color in the genome following LAI. Image sourced from https://gnomad.broadinstitute.org/news/2021-12-local-ancestry-inference-for-latino-admixed-american-samples-in-gnomad/
Within the realm of accomplishing the goals of a genome wide association test – to see if a trait of interest is associated with a certain genetic variant – local ancestry inference is especially helpful in identifying genetic variants associated with traits or diseases in admixed groups, or ethnic groups whose genomes have resulted from a recent mixture of two or more geographically distinct ancestral populations. In short, local ancestry is particularly useful in genetic analyses of diverse cohorts, in addition to being useful for admixture mapping. Some admixed populations of interest include African Americans with African and European Ancestry and Latino Americans who have African, European, and Native American ancestry. In summary, genetic admixture mapping using LAI has interdisciplinary applications, as it can allow for a richer understanding of historical human migration to further corroborate current historical analyses using GWAS.